전문의약품, 제네릭의약품, 의료기기 모두 전년 대비 인상, 바이오시밀러만 인하
전문의약품 신약 신청 시 드는 비용은 사상 처음 400만 달러 넘어

[바이오타임즈] 미국 식품의약국((Food and Drug Administration, FDA)의 2024년 회계연도 전문의약품 신약 신청 비용이 사상 최초로 400만 달러(약 53억 원)를 넘어설 것으로 보인다. 지난 2022년도 회계연도 기준 300만 달러에서 2년 만에 100만 달러(약 13억 원)이 늘어난 것이다.
한국바이오협회 바이오경제센터의 자료에 따르면, 오는 10월부터 적용되는 미국 FDA의 2024년 회계연도 의약품, 의료기기 심사 수수료가 확정됐다. FDA는 매년 인플레이션, 심사 신청건 수, 제조시설 수 등을 고려해 이용자인 기업으로부터 받는 허가심사 수수료를 책정하고 있다.
미국 FDA는 2023년 7월 28일, 신약(전문의약품), 제네릭, 바이오시밀러, 의료기기 제조기업으로부터 받는 2024년 회계연도 허가심사 수수료(User fee)를 확정해 발표했다. 2024년도 회계연도는 2023년 10월부터 2024년 9월까지 적용된다.
전문의약품을 비롯해, 제네릭 의약품, 의료기기 모두 전년 대비 인상됐으며, 특히 전문의약품 신약 신청 시 드는 비용은 사상 처음 400만 달러가 넘었다. 반면 바이오시밀러의 허가심사 수수료는 큰 폭으로 감소할 예정이다.
올해 10월 1일부터 적용되는 전문의약품에 대한 2024년도 허가심사 수수료는 4,048,695달러(약 52억 9,100만 원)로 책정되어 2023년에 비해 24.9% 인상될 예정이다. 제네릭 의약품의 허가심사 수수료는 4.9% 인상된 252,453달러(약 3억 3,100만 원), 의료기기는 사전 허가·사전 신고·신기술 모두 9.5% 인상된 각 483,560달러(약 6억 3,500만 원), 21,760달러(약 2,900만 원), 145,068달러(약 1억 9,000만 원)이다.
바이오시밀러 허가심사 수수료는 절반 가까운 41.7%가 인하된 1,018,753달러(약 13억 3,700만 원, 임상자료 포함 시)이다. 이러한 인하가 가능한 요인으로는 2023년에서 이월된 약 2,000만 달러의 바이오시밀러 운영비가 2024년 예산에 반영되면서 허가심사비용이 크게 낮아졌기 때문으로 분석된다.
허가심사 수수료(이용자부담금)란 기업들이 FDA에 의약품이나 의료기기 등의 시판 허가 등을 위해 내야 하는 비용으로, 의약품이나 의료기기의 종류별로 비용에 차이가 있다. 중소기업이나 희귀의약품 등의 여부에 따라 비용은 면제 또는 감면된다.
의약품이나 의료기기는 시장에 출시되기 전에 여러 단계의 복잡한 공급 사슬을 거치고, 점점 관련된 인허가 규제 업무가 복잡해지고 새로운 규제 환경이 도입되어야 하는 현실 등이 반영 되면서 심사 수수료도 증가 추세이다.
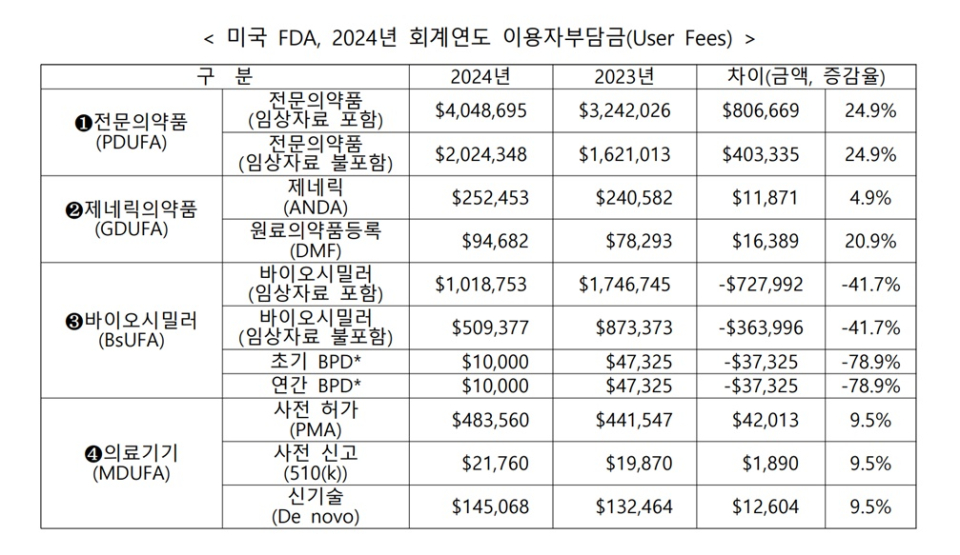
전문의약품의 이용자부담금(심사 수수료)는 전문의약품 이용자부담금법(PDUFA: Prescription Drug User Fee Act)에 따라 정해지며, 임상시험 자료 검토 필요 여부에 따라 필요한 경우, 필요 없는 경우보다 비용이 2배나 높다.
제네릭 의약품의 이용자부담금은 제네릭 의약품 이용자부담금개정법(GDUFA: Generic Drug User Fee Amendments)에 따라 정해지며, 원료의약품 또는 완제의약품 시설이나 해외나 국내 소재 여부에 따라 비용 차이가 발생한다.
바이오시밀러의 이용자부담금은 바이오시밀러 이용자부담금개정법(BsUFA: Biosimilar User Fee Amendments)에 따라 정해지며, 전문의약품과 마찬가지로 임상자료 심사 필요 여부에 따라 비용에 있어 2배 차이가 난다.
BPD(Biological product development)는 바이오시밀러의 개발 및 승인을 신속하게 진행시키기 위한 미국 FDA 심사관과 기업과의 공식적인 미팅 프로그램으로, 초기(Initial BPD)와 연간(Annual BPD)으로 구분된다.
의료기기의 경우, 의료기기 이용자부담금개정법(MDUFA: Medical Device User Fee Amendments)에 따라 정해지며, 사전 허가나 사전신고가 필요한 경우나 신기술(De novo) 의료기기 등에 따라 비용 차이가 발생한다.
한편 미국 FDA는 미국에서 생산, 유통, 판매되는 모든 종류의 의약품 및 의료기기에 대해 세부 법령에 규정하여 통제, 관리, 승인을 담당한다(의료기기는 Class I, II, II로 분류). 의약품과 의료기기가 대상인 ‘허가 및 승인’은 환자나 일반인을 대상으로 임상 1상부터 3상까지 진행하면서 의약품과 의료기기의 안전성과 효능을 입증해야 한다.
최근에는 각 기업이 별도의 지정이 필요 없으며, 심사가 빠르게 진행되는 신속 프로그램을 활용하는 경우가 증가하고 있다. FDA의 신속 심사제도는 크게 ▲패스트 트랙(Fast Track) ▲혁신의약품 지정(Breakthrough Therapy Designation) ▲가속 승인(Accelerated Assessment) ▲우선심사(Priority Review) 등 4가지가 있다.
하지만 가속 승인의 경우, 무분별한 허가로 실효성이 제기되고 있다. 가속 승인제도는 중증질환이나 아직 치료제가 개발되지 않은 의료상 요구가 많은 질환을 대상으로 신약후보물질들에 대해 임상시험이 끝나기 전에 조건부 허가를 내주는 제도이다. 특히, FDA가 시중에서 약물을 철회할 강력한 권한이 없기 때문에 후속 연구 결과 확인이 어려워 여러 가지 지적을 받아왔다.
이러한 영향 탓으로 지난해 미국 식품의약국(FDA)의 신약 허가는 6년 만에 최저치를 기록했다. FDA 약품평가연구센터에 따르면 ▲신물질 신약(NME) 22개 ▲바이오신약(BLA) 15개 등 총 37개 신약이 허가됐다. 백신, 세포 및 유전자치료제, 혈액제제 등은 포함되지 않은 수치다. 이는 2021년 허가 시 논란이 됐던 알츠하이머 치료제 ‘아두헬름(성분명: 아두카누맙)’의 영향으로 심사가 강화된 것으로 보인다.
[바이오타임즈=김수진 기자] sjkimcap@biotimes.co.kr
