기존 방법 대비 2배 향상된 스크리닝 성공률 달성
단백질-리간드 간 상호작용 예측 통해 약물 설계 등에 적용 기대

[바이오타임즈] 신약 개발에 드는 기간은 평균 10년, 비용은 1조 원에 달한다. 하지만 1만 개의 후보 약물 중 개발에 성공해 시판까지 가는 경우는 1개에 불과할 정도로 성공확률도 희박하다.
신약 개발에 인공지능을 활용하면 수많은 화합물을 반복해서 합성·실험하는 전통적 방법에서 벗어나 시간과 비용을 대폭 절감할 수 있다. 또한, 파이프라인 확대도 가능하다.
인공지능으로 약물 후보 분자를 발굴하기 위해서는 타깃 단백질과 강하게 결합하는 리간드를 찾는 것이 중요하다.
리간드란 수용체와 같은 큰 생체 분자에 특이적으로 결합하는 물질을 말하며, 생체 내의 중요한 요소이자 의약품의 개발 등에 큰 역할을 한다. 단백질-리간드 결합에서 리간드는 일반적으로 표적 단백질의 특정 부위에 결합하여 신호를 발생시키는 분자로, 이러한 결합은 보통 표적 단백질의 입체 구조 변화를 초래한다.
하지만 유효 물질을 찾기 위해 수백만에서 수천만 개의 무작위 리간드 라이브러리를 대상으로 실험 전수 조사를 수행하는 것은 천문학적인 시간과 비용이 필요하다. 이러한 시간과 비용을 절감하기 위해 최근 단백질-리간드 상호작용 예측에 기반한 가상탐색(Virtual Screening) 기술이 주목받고 있다.
가상탐색은 통계적 혹은 기계학습 모델을 이용해 약물-타깃 상호작용을 빠르게 예측한다. 가상탐색 결과에서 약물-타깃 상호작용이 좋지 않은 경우를 제거한 후 신약으로 가능성이 큰 활성 조합만을 다음 단계인 고속 대량 스크리닝 과정으로 보내 신약 개발 과정의 비용과 시간을 크게 줄일 수 있도록 도와준다.
그런데 기존의 상호작용 예측 인공지능 모델들은 학습에 사용한 구조에 대해서는 높은 예측 성능을 보여주지만, 새로운 단백질 구조에 대해서는 낮은 성능을 보이는 과적합(over-fitting)이 문제가 됐다. 과적합 문제는 일반적으로 모델의 복잡도에 비해 데이터가 적을 때 발생한다.
이러한 과적합 문제를 해결함으로써 다양한 단백질에 대해 고른 성능을 보여주는 예측 모델을 국내 연구진이 개발했다.
◇카이스트 김우연 교수팀, 일반화 성능 높인 단백질-리간드 상호작용 예측 모델 개발
KAIST는 화학과 김우연 교수 연구팀이 교원창업 인공지능 신약 개발 스타트업 HITS 연구진과 함께 물리 기반 삼차원 그래프 심층 신경망을 이용해 일반화 성능을 높인 단백질-리간드 상호작용 예측 모델을 개발했다고 17일 밝혔다.
한국연구재단의 지원을 받아 수행된 이번 연구는 국제 학술지 ‘Chemical Science(IF=9.825)’ 2022년 4월 13호에 표지 논문 및 ‘금주의 논문(Pick of the Week)’으로 선정됐다. (논문명: PIGNet: a physics-informed deep learning model toward generalized drug–target interaction predictions, 논문 링크: https://doi.org/10.1039/D1SC06946B)
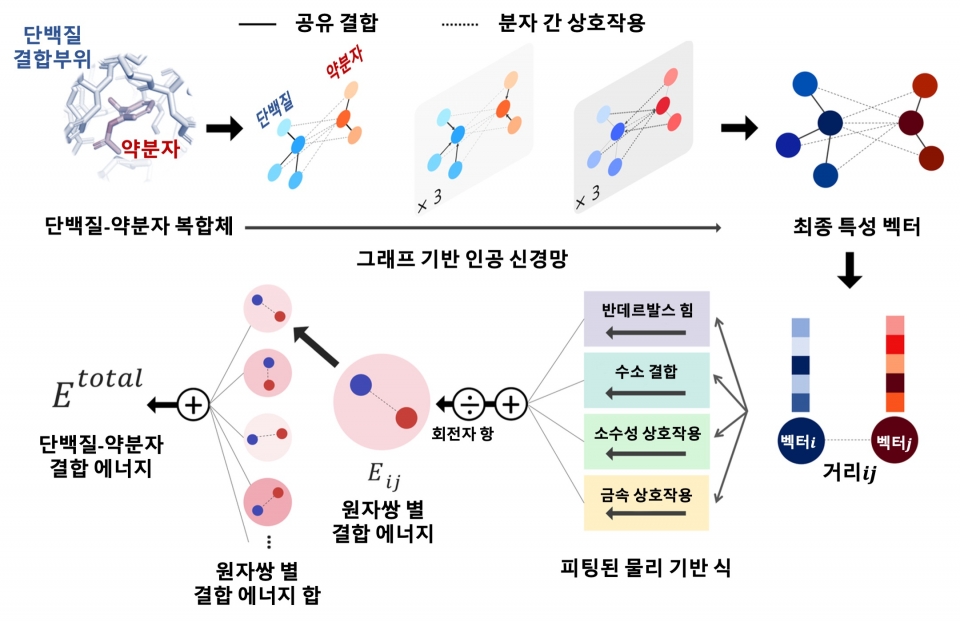
연구진은 물리화학적 아이디어들을 딥러닝 모델에 적용해 모델의 복잡도를 줄이면서 물리 시뮬레이션을 통해 부족한 데이터를 보강함으로써 과적합 문제를 해결하고자 했다. 단백질 원자와 리간드 원자 사이의 거리에 따른 반데르발스 힘, 수소 결합력 등을 물리화학적 방정식으로 모델링하고, 매개변수를 딥러닝으로 예측함으로써 물리 법칙을 만족하는 예측을 가능하게 했다.
또한, 학습에 사용한 단백질-리간드 결정 구조가 실험적으로 판명된 가장 안정한 구조임에 착안했다. 부족한 실험 데이터를 보강하기 위해 불안정한 단백질-리간드 구조로 이루어진 수십만 개의 인공 데이터를 생성해 학습에 활용했고, 그 결과 생성된 구조에 비해 실제 구조를 안정하게 예측하도록 모델을 학습할 수 있었다.
◇기존 방법 대비 2배 향상된 스크리닝 성공률 달성
연구진은 개발된 모델의 성능을 검증하기 위해 대조군으로 ‘CASF-2016 벤치마크’를 활용했다. 이 벤치마크는 다양한 단백질-리간드 구조들 사이에서 실험적으로 판명된 결정 구조에 근접한 구조를 찾는 도킹과 상대적으로 결합력이 큰 단백질-리간드 쌍을 찾는 스크리닝 등 실제 약물을 개발하는 과정에 필수적인 과제를 포함하고 있다.
검증 테스트 결과 기존에 보고된 기술에 비해 높은 도킹 및 스크리닝 성공률을 보여줬으며, 특히 스크리닝 성능은 기존에 보고된 최고 성능 대비 약 두 배 높은 수치를 보였다.
연구진이 개발한 물리 기반 딥러닝 방법론의 또 다른 장점은 예측의 결과를 물리적으로 해석 가능하다는 것이다. 이는 딥러닝으로 최적화된 물리화학 식을 통해 최종 상호작용 값을 예측하기 때문이다. 리간드 분자 내 원자별 상호작용 에너지의 기여도를 분석함으로써 어떤 작용기가 단백질-리간드 결합에 있어서 중요한 역할을 했는지 파악할 수 있으며, 이와 같은 정보는 추후 약물 설계를 통해 성능을 높이는 데 직접 활용할 수 있다.
공동 제1 저자로 참여한 KAIST 화학과 문석현, 정원호, 양수정(현재 MIT 박사과정) 박사과정 학생들은 “데이터가 적은 화학 및 바이오 분야에서 일반화 문제는 항상 중요한 문제로 강조돼왔다ˮ며 ”이번 연구에서 사용한 물리 기반 딥러닝 방법론은 단백질-리간드 간 상호작용 예측뿐 아니라 다양한 물리 문제에 적용될 수 있을 것ˮ이라고 말했다.
[바이오타임즈=김수진 기자] sjkimcap@biotimes.co.kr
